(Kopie 1)
Despite their aggressiveness and dismal prognosis, pediatric sarcomas often exhibit an oligo-mutated genomic signature. Among these mutations however, pathognomonic and unique fusion-genes with strong transformative effects, such as EWSR1::FLI1 (Ewing sarcoma) or PAX3::FOXO1 (alveolar rhabdomyosarcoma), are found in many bone and soft tissue cancers of children and adolescents. Notably, many of these fusion oncogenes drive malignant transformation by acting as aberrant transcription factors.
While the discovery of these fusion genes has greatly improved precise molecular diagnosis of their respective entity, no targeted therapies have been developed regardless of their ideal nature as tumor-specific, pharmacological targets.
In this proof-of-concept study, we took advantage of the neomorphic DNA binding properties of EWSR1::ETS fusion genes, which constitute the most common fusion oncogenes in Ewing sarcoma. For example, while wildtype FLI1 and ERG bind DNA at ETS-like motifs with a GGAA core-motif, EWSR1::ETS binds to repetitive GGAA-motifs, i.e. GGAA-microsatellites (mSats), which are thereby converted into potent neoenhancers.
By fusing GGAA-repeats to a synthetic minimal promoter, we accomplished tumor-specific gene expression of various therapeutic transgenes, such as herpes simplex thymidine kinase (suicide gene), luciferase (reporter gene) and XCL-1 and IL-15 (immunomodulating cytokines). By coupling this expression cassette with a GPR64-targeted transduction strategy (a surface antigen, highly and specifically expressed in Ewing sarcoma), we were able to achieve tumor regression in vivo without noticeable side effects. Moreover, we successfully generated and tested a synthetic expression cassette for PAX3::FOXO1-positive alveolar rhabdomyosarcoma.
In summary, our study proves the strength of novel, targeted strategies in the fight against pediatric sarcoma by exploiting, rather inhibiting neomorphic functions of fusion oncogenes in these tumor types. (pubmed)
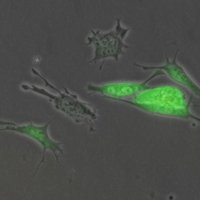
The identification of cancer stemness genes is crucial to understand the underlying biology of therapy resistance, relapse, and metastasis. In pediatric tumors, mutations generally do not involve canonical stemness/metastasis-associated genes.
Ewing sarcoma (EwS), the second most common bone cancer in children and adolescents, is a highly aggressive malignancy associated with dismal outcome in metastatic disease, wherefore deciphering mechanisms of metastasis is imperative.
EwS is characterized by a remarkably silent genome with a single driver mutation that promotes stem cell features. Thus, EwS constitutes an ideal model to study how perturbation of a transcriptional network by a dominant oncogene can mediate metastasis.
In this study, we took a fresh approach to explore mechanisms of metastasis by investigating how the EWSR1-FLI1 master oncogene perturbs transcriptional networks and identified TCF7L1 as a key hub of metastasis-associated gene networks.
By integrating omics-analyses and functional experiments, we demonstrated that TCF7L1 is a critical mediator of metastasis in EwS, which may be utilized as a prognostic biomarker.
In summary, this study exemplifies the power of systems biology to decipher gene regulatory networks and to identify key players in the metastatic process, which may be highly relevant for and translatable to other oligomutated (pediatric) cancers. (pubmed)

Chromosomal instability (CIN) is a hallmark of cancer. Through moderate CIN, most cancer types acquire intra-tumor heterogeneity that enhances the overall 'fitness' of the cancer cell population. Yet, excessive CIN can lead to non-viable karyotypes. While CIN-induction in cancer types that are intrinsically genomically instable may have little therapeutic benefit, it may cause massive cell death in cancers with 'silent' or nearly diploid genomes, such as in many pediatric cancer types including Ewing sarcoma (EwS) – the second most common bone or soft-tissue cancer in children and adolescents. This study showed that CIN-induction, e.g., by uncoupling mitosis and cytokinesis, may constitute a new therapeutic option for EwS.
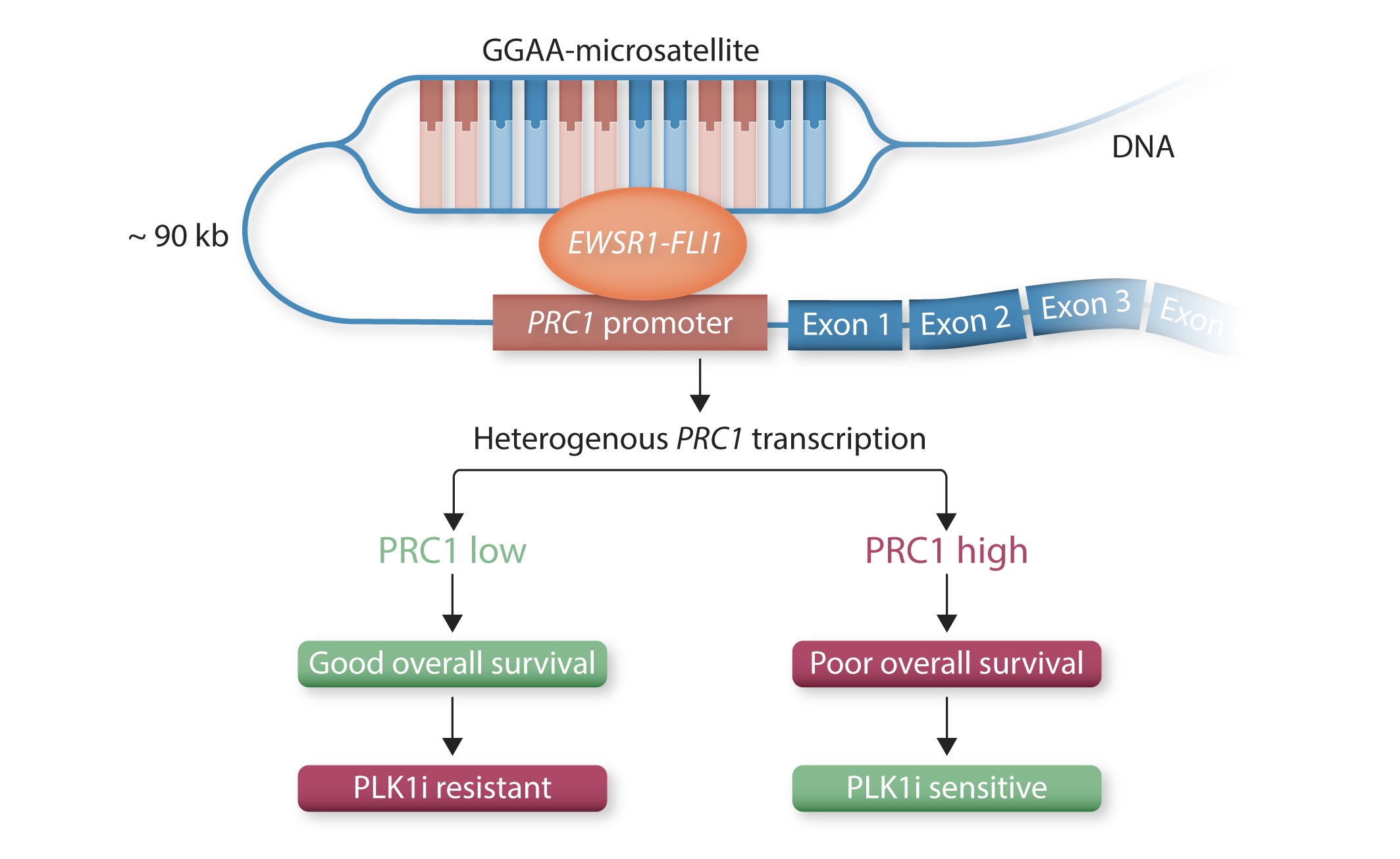
To identify a candidate gene that could offer a large therapeutic window, we analyzed comprehensive gene expression microarray datasets, in which protein regulator of cytokinesis 1 (PRC1) exhibited the greatest fold change among significantly overexpressed cytokinesis-related genes in EwS compared to normal tissues. High PRC1 expression was correlated with poor patient outcome and proved to constitute an independent risk-factor for EwS patients. Through integration of time-course in vitro and in vivo gene expression analyses, DNase-seq and ChIP-seq data, as well as CRISPR-mediated enhancer editing and 3C-PCR assays, we found that EWSR1-FLI1 directly hijacks PRC1, which physiologically safeguards controlled cell division, through binding to a proximal enhancer-like GGAA-microsatellite. By combing transcriptome-wide gene expression and analyses with in vitro and in vivo RNA interference experiments we then demonstrate that PRC1 overexpression induces a highly proliferative phenotype in EwS cells while ensuring proper cell division. Interestingly, high PRC1 expression creates a therapeutic vulnerability toward PLK1 inhibition that can repress growth of even chemo-resistant EwS cells by triggering mitotic catastrophe, which can be fully ablated by genetic deletion of the PRC1-associated GGAA-microsatellite enhancer. In sum, our data suggested that PRC1 may serve as a promising predictive biomarker for therapies causing cytokinesis defects and unviable karyotypes in genomically 'silent' tumors. More importantly, PRC1-related mechanism identified in our EwS model may be translatable to other cancers for which detection of high PRC1 could serve as a predictive biomarker, our discovery has the potential to initiate a clinical renaissance of PLK1i in many cancer types. (pubmed)
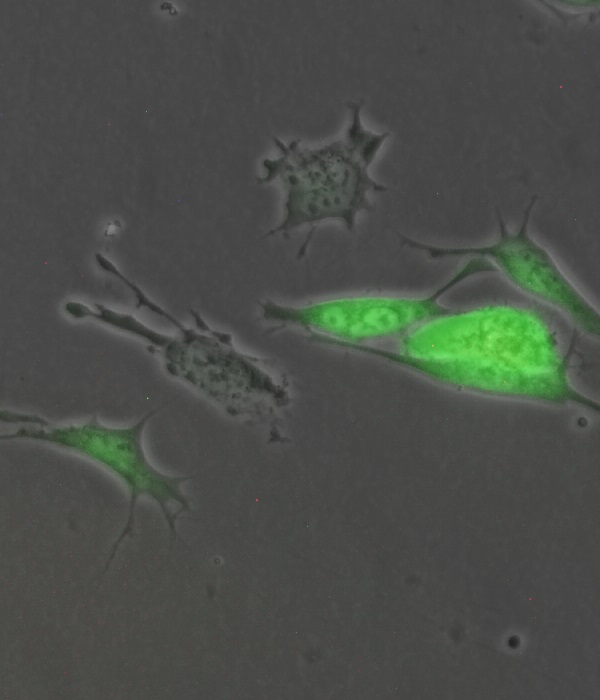
Ewing sarcoma (EwS) is a highly aggressive bone- or soft tissue-associated cancer in children, adolescents, and young adults, for which the development of more effective therapies remains an unmet medical need. This study not only sheds light on a critical role of RRM2, a small subunit of the ribonucleotide reductase, which catalyzes the rate limiting de novo DNA synthesis, for aggressive phenotypes, but also demonstrated that RRM2 can be a clinically actionable target in EwS.
Employing a multi-layered screening approach, we carefully selected candidate genes, which are highly expressed in EwS but not in normal tissues, whose overexpression potentially confers negative clinical outcome, and for which a selective drug and pharmacokinetic data were available. In this screen, we identified a single putative druggable target, RRM2, that fulfilled these criteria. Using transcriptome and matched datasets of clinical tumor samples, we showed that RRM2 mRNA and protein overexpression is associated with an aggressive phenotype such as early metastasis or disease recurrence and thus poor overall survival of patients, suggesting RRM2 as potential prognostic biomarker for EwS. In agreement with these clinical findings, RRM2 silencing as well as its pharmacological inhibition with the specific inhibitor triapine (3-AP) significantly reduced EwS growth in vitro and in vivo. Interestingly, triapine could overcome chemoresistance against doxorubicin or gemcitabine, and synergize with cell cycle checkpoint inhibitors for CHEK1 or WEE1 in cell models. Collectively, this study provided a translational rationale to exploit RRM2 as a novel therapeutic target in EwS, encouraging further (pre)clinical investigations. (pubmed)
